

「クロマトグラフィー分析」、「クロマトグラフィー」としても知られるクロマトグラフィーは、分析化学、有機化学、生化学、その他の分野で非常に広範囲に応用される分離および分析方法です。
クロマトグラフィーの創始者はロシアの植物学者M.ツヴェッターです。1906年、ロシアの植物学者ズヴェッターは、植物色素を分離するために、植物色素を含む石油エーテル抽出物を炭酸カルシウム粉末の入ったガラス管に注ぎ、石油エーテルで上から下に溶出させた実験結果を発表しました。顔料が異なると炭酸カルシウム粒子の表面に吸着能力が異なるため、浸出の過程で顔料が異なる速度で下に移動し、その結果、異なる色の帯が形成されます。顔料成分を分離した。彼はこの分離方法をクロマトグラフィーと名付けました。
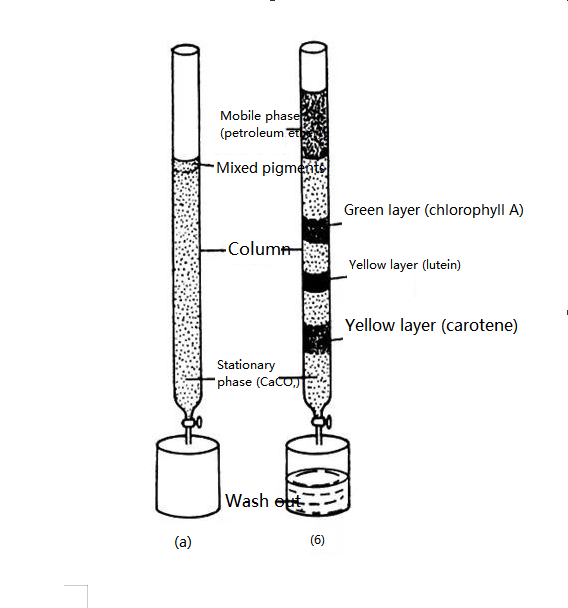
植物葉色素分離実験の模式図
分離方法の継続的な発展に伴い、分離の対象となる無色の物質が増え、クロマトグラフィーも徐々に「色」の意味を失いましたが、その名前は現在でも使用されています。
クロマトグラフィーによる分類
クロマトグラフィーの本質は、分離される分子が固定相と移動相の間で分配され、バランスが保たれるプロセスです。異なる物質は 2 相間で異なる方法で分配されるため、移動相では異なる速度で移動します。移動相の移動により、混合物中のさまざまな成分が固定相上で互いに分離されます。メカニズムに応じて、さまざまなカテゴリに分類できます。
1、二相物理状態分類による
移動相:ガスクロマトグラフィー、液体クロマトグラフィー、超臨界流体クロマトグラフィー
固定相: 気体-固体、気体-液体。液体-固体、液体-液体
2、固定相分類の形式による
カラムクロマトグラフィー: パックドカラムクロマトグラフィー、キャピラリーカラムクロマトグラフィー、マイクロパックドカラムクロマトグラフィー、分取クロマトグラフィー
平面クロマトグラフィー:ペーパークロマトグラフィー、薄層クロマトグラフィー、高分子膜クロマトグラフィー
3、分離メカニズムによる分類
吸着クロマトグラフィー: 吸着剤への吸着および脱着能力に応じて、さまざまな成分を分離します。
分配クロマトグラフィー: 溶媒への溶解度に応じてさまざまな成分を分離します。
分子排除クロマトグラフィー: 分離の分子サイズに応じて イオン交換クロマトグラフィー: イオン交換樹脂分離における親和性の異なる成分
アフィニティークロマトグラフィー:生体高分子間の特異的親和性の存在を利用した分離
キャピラリー電気泳動: 移動度および/または分配挙動の違いに従って成分が分離されました
キラルクロマトグラフィーは、キラル薬物の分離と分析に使用されます。キラルクロマトグラフィーは、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法、キラル誘導体化試薬法の 3 つに分類できます。キラル移動相添加法;キラル固定相分割法
クロマトグラフィーの基本用語
クロマト分離検出後の各成分の応答信号を時間に対してプロットした曲線をクロマトグラムと呼びます。
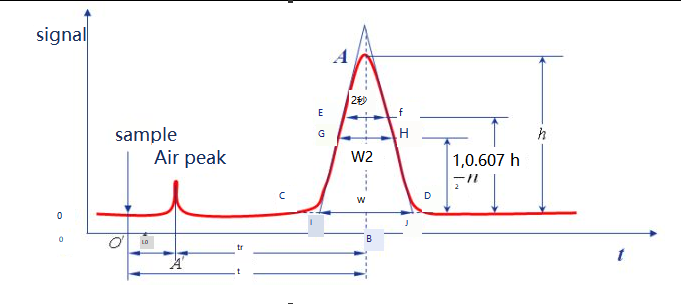
ベースライン:特定のクロマトグラフィー条件下で、移動相のみが検出器システムを通過するときに生成される信号の曲線は、OT 線に示すようにベースラインと呼ばれます。実験条件が安定している場合、ベースラインは水平軸に平行な線でした。ベースラインは、時間の経過に伴う機器、主に検出器のノイズを反映します。
ピークの高さ:クロマトグラフィーのピーク点とベースラインの間の垂直距離。AB' 線で示される h で示されます。
領域幅:クロマトグラフィーのピークの領域幅は、分離効率に直接関係します。クロマトグラフィーのピーク幅を記述するには、標準偏差 σ、ピーク幅 W、および FWHM W1/2 の 3 つの方法があります。
標準偏差 (σ) :σ は正規分布曲線上の 2 つの変曲点間の距離の半分であり、σ の値は列から離れる成分の分散度を示します。σの値が大きいほど排水成分が分散し、分離効果が悪くなります。逆に排水成分は濃縮されており、分離効果は良好です。
ピーク幅W:クロマトグラフィーピークの両側の交点は接線として使用され、ベースライン上の切片はピーク幅またはベースライン幅と呼ばれ、図1Jに示すようにWで表すこともできます。正規分布の原理によれば、ピーク幅と標準偏差の関係は W=4σ であることが証明されます。
W1/2:GH の距離で示されるように、ピーク高さの半分のピーク幅は FWHM と呼ばれます。W1/2=2.355σ、W=1.699W1/2。
W1/2、W は両方とも σ から導出され、カラム効果の測定に加えてピーク面積を計算するために使用されます。FWHM 測定はより便利であり、最も一般的に使用されています。
簡単な概要
クロマトグラフィーのピーク流出曲線から、次の目的を達成できます。
a, クロマトグラフィーのピークの保持値に基づいて定性分析を実行しました。
b、クロマトグラフィーのピークの面積またはピークに基づく定量分析
カラムの分離効率は、クロマトグラフィーピークの保持値とピーク幅に基づいて評価されました。
クロマトグラフィーの計算式
1. 保持値
保持値は、サンプル成分がカラム内に保持される程度を表すために使用されるパラメータであり、クロマトグラフィーの特性評価の指標として使用されます。その表現方法は以下の通りです。
保持時間 tR
死亡時刻tM
保持時間 tR を調整する'=tR-tM
(定常期に費やした合計時間)
保持量
VR=tR*F.(移動相速度に依存しない)
デッドボリューム
VM=tM*Fc
(注入器から検出器までの流路のうち、固定相が占有していない空間)
リテンションボリュームVRを調整する'=t'R*FC
2. 相対保持値
相対保持値は、分離係数、分配係数比、または相対容量係数とも呼ばれ、特定のクロマトグラフィー条件下での、試験対象成分の調整された保持時間 (体積) と標準物質の調整された保持時間 (体積) の比です。

相対保持値は、流量や固定剤の損失などの特定の操作条件が保持値に及ぼす影響を排除するために使用されました。相対保持値の基準は、試験サンプル中の成分または人為的に添加された化合物とすることができます。
3. リテンションインデックス
保持指数は、固定溶液 X 中での被検物質 i の保持指数です。参照物質として炭素数 N と N+n の 2 つの n-アレーンを選択します。それらの調整された保持時間はそれぞれ t 'r (N) と t 'r (N+n) であるため、被験物質 i の調整された保持時間 t 'r (i) はちょうどそれらの間にあります。てーる(N)。
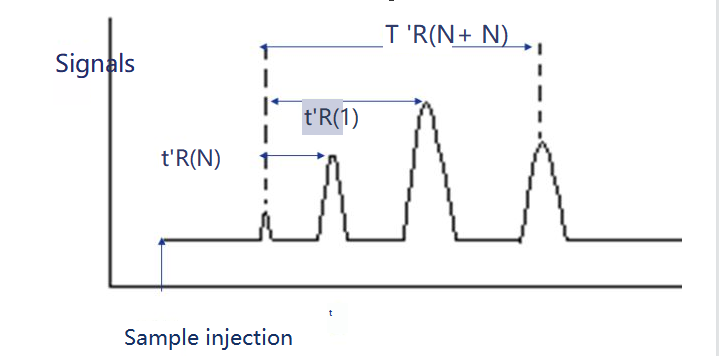
保持指数は次のように計算できます。
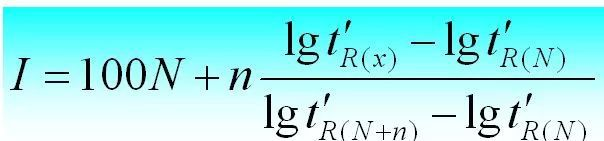
4. 設備利用率(k)
平衡状態における、固定相の成分の質量 (s) と移動相 (m) の比であり、容量係数と呼ばれます。式は次のとおりです。
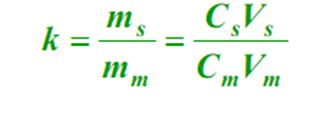
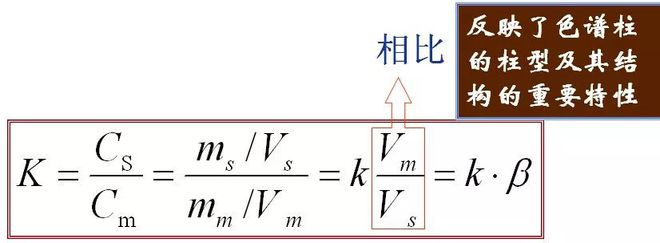
5、分配係数(K) 平衡状態における固定相成分の濃度(s)と移動相(m)の濃度の比を分配係数といいます。式は次のとおりです

K と k の関係:
柱のタイプとその結び目の重要な構造特性を反映します。
簡単な概要
保持値と設備利用率および分配係数との関係:
クロマトグラフィーによる分離は、固定された相対サンプル中の各成分の吸着または溶解能力の違いに基づいており、これは分配係数 K (または容量係数 k) 値の大きさによって定量的に表すことができます。
吸着力や溶解力が強い成分は分配係数(容量係数)が大きく、保持時間が長くなります。逆に、吸着力や溶解度が弱い成分は分配係数が小さく、保持時間が短くなります。
クロマトグラフィーの基礎理論
1. トレイ理論
(1) 熱力学理論の提唱
MartinとSingeが提案したタワープレートモデルから始まりました。
分留塔: 異なる分離の沸点に応じて、トレイ内で気液平衡を数回行います。
カラム: 成分は 2 相間の複数の分配によってバランスがとられ、異なる分配係数に従って分離されます。
(2) 仮説
(1) カラム内には多くのトレイがあり、成分はトレイ間隔 (つまりトレイの高さ) 内ですぐに分布平衡に達します。
(2) 移動相は連続的にではなく脈動してカラムに入ります。つまり、各通過がカラムの体積となります。
(3) サンプルを各カラムプレートに添加した場合、カラム軸に沿ったサンプルの拡散は無視できます。
(4) 分配係数は、成分の量に関係なく、すべてのトレイで同じです。つまり、分配係数は各タバンで一定です。
(3) 原則
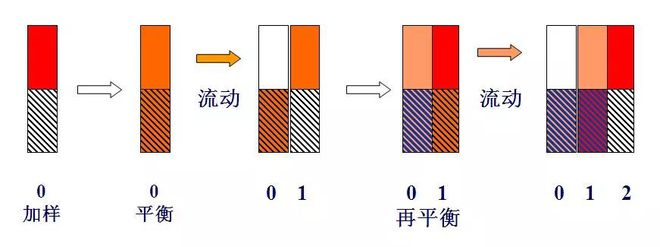
トレイ理論の模式図
単位質量の成分、すなわちm=1(例えば、1mgまたは1μg)を0番トレーに加え、分配平衡後、k=1、すなわちns=nmであるため、nm=ns=0.5となる。
プレート体積(lΔV)のキャリアガスが脈動してプレート0に入ると、気相中のnm成分を含むキャリアガスがプレート1に押し出されます。このとき、プレート0の液相中のns成分は、そしてプレート 1 の気相中の nm 成分は 2 つの相間で再分配されます。したがって、プレート 0 に含まれる成分の総量は気相と液相がそれぞれ 0.25 ずつ 0.5 となり、プレート 1 に含まれる成分の総量も 0.5 になります。気相と液相も 0.25 でした。
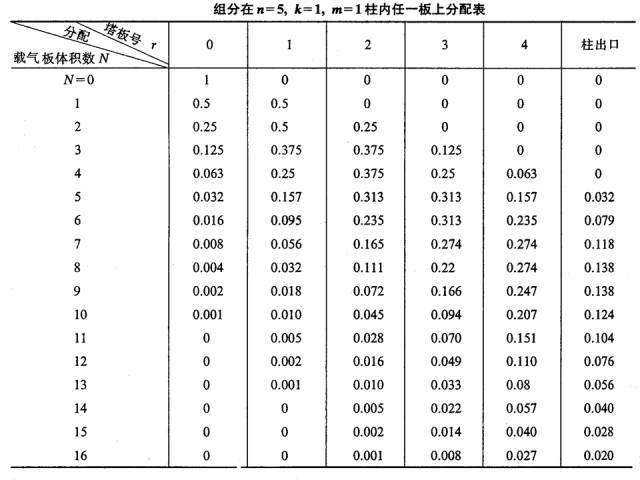
このプロセスは、新しいプレートボリュームのキャリアガスがカラム内に脈動されるたびに繰り返されます (以下の表を参照)。
(4)クロマトグラフの流出曲線式

σ は標準偏差、 は保持時間、C は任意の時点の濃度、
C は注入濃度、つまり成分の総量 (ピーク面積 A) です。
(5) カラム効率パラメータ
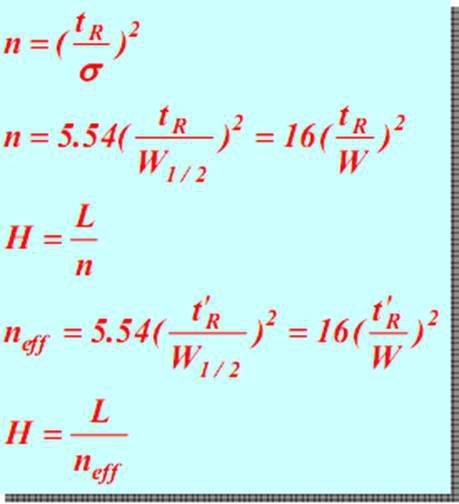
一定の tR では、W または w 1/2 が小さいほど (つまり、ピークが狭いほど)、理論段数 n が大きくなり、理論段高さが小さくなり、カラムの分離効率が高くなります。有効理論トレイネフについても同様です。したがって、理論トレイ数はカラムの効率を評価する指標となります。
(5)特徴と欠点
> 利点
トレイ理論は半経験的であり、流出曲線の形状を説明します。
コンポーネントの分割および分離プロセスが図示されています
カラムの効率を評価する指標を提案
> 制限事項
次の 2 つの段階では、コンポーネントは実際には分布平衡に達することはできません。
カラム内の成分の長手方向の拡散は無視できません。
物質移動プロセスに対するさまざまな速度論的要因の影響は考慮されていません。
カラム効果と移動相の流速の関係は説明できません。
どのような主な要因が柱効果に影響を与えるかは不明です
これらの問題はレート理論で十分に解決されます。
2. レート理論
1956 年、オランダの学者 VanDeemter らは、はトレイ理論の概念を吸収し、トレイの高さに影響を与える速度論的要因を組み合わせ、クロマトグラフィープロセスの速度論的理論、つまり速度理論を提唱し、VanDeemter 方程式を導き出しました。それは、クロマトグラフィープロセスを動的非平衡プロセスとみなして、ピークの広がりに対する速度論的要因の影響 (つまり、カラム効果) を研究します。
その後、ギディングスとスナイダーらは、は、VanDeemter 方程式 (後にガスクロマトグラフィー速度方程式と呼ばれる) に基づいて、液体と気体の性質の違いに従って、液体クロマトグラフィー速度方程式 (すなわち、Giddings 方程式) を提案しました。
(1) ファン・ディームター方程式
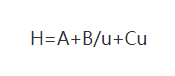
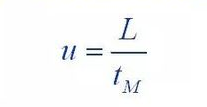
ここで、 H: ボードの高さです。
A: 渦拡散項の係数
B: 分子拡散項の係数
C: 物質移動抵抗項の係数
(2) ギディングス方程式
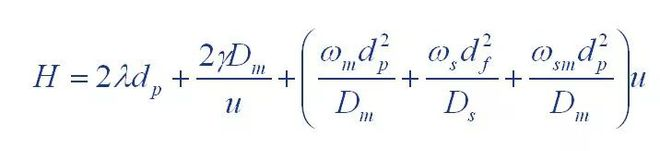
定量的分析と定性的分析
(1) 定性分析
定性クロマトグラフィー分析は、各クロマトグラフィーのピークによって表される化合物を決定することです。さまざまな物質は特定のクロマトグラフィー条件下で明確な保持値を示すため、保持値は定性的な指標として使用できます。現在、さまざまなクロマトグラフィー定性法は保持値に基づいています。
ただし、異なる物質は、同じクロマトグラフィー条件下で類似または同一の保持値を有する場合があります。つまり、保持値は排他的ではありません。したがって、保持値のみに基づいて完全に未知のサンプルの特性を評価することは困難です。サンプルの出所、性質、目的の理解に基づいてサンプルの組成を予備的に判断でき、次の方法を使用してクロマトグラフィーのピークで表される化合物を決定できます。
1. 純物質を用いた定性管理
特定のクロマトグラフィー条件下では、未知の物質の保持時間は定義されています。したがって、同じクロマトグラフィー条件下での既知の純粋な物質の保持時間を未知の物質の保持時間と比較することによって、未知のものを定性的に同定することができます。両者が同じであれば、未知の物質は既知の純粋な物質である可能性があります。そうでなければ、未知のものは純粋な物質ではありません。
純物質管理法は、組成が既知で、組成が比較的単純で、純物質が既知である未知物質にのみ適用できます。
2. 相対保持値法
相対保持値 α は、成分 i と標準物質の間の調整を指します。保持値の比率:
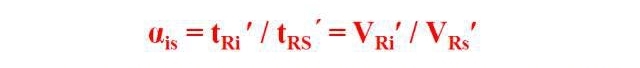
固定液とカラム温度の変化によってのみ変化し、他の操作条件とは関係ありません。
特定の固定相とカラム温度で、成分 i と標準物質 s の調整後の保持値がそれぞれ測定され、上式に従って計算されます。得られた相対保持値は、文献内の対応する値と定性的に比較できます。
3、既知の物質を追加してピーク高さを高める方法
未知試料の成分が多い場合や、未知試料が特定項目の分析のみに使用される場合、得られるクロマトグラフィーのピークが密すぎて上記の方法では識別しにくい場合があります。
「まず未知のサンプルのクロマトグラムが作成され、次に未知のサンプルに既知の物質を添加することによってさらなるクロマトグラムが得られます。」このような物質では、ピーク高さが増加した成分を知ることができます。
4. 指数の定性的手法を維持する
保持指数は、固定剤上の物質の保持挙動を表し、現在 GC において最も広く使用され、国際的に認知されている定性指数です。再現性が良く、標準が均一で、温度係数が小さいという利点があります。
保持指数は固定相の特性とカラム温度のみに関係し、他の実験条件には関係しません。その精度と再現性は優れています。カラム温度が固定相の温度と同じである限り、文献値を同定に適用でき、比較のために純粋な物質を使用する必要はありません。
(2)定量分析
クロマトグラフィー定量の基礎:
定量分析の仕事は、混合サンプル中の何百もの成分を見つけることです
断片的なコンテンツ。クロマトグラフィーによる定量化は、以下に基づいて行われました: 操作条件が一貫している場合、
測定された成分の質量(または濃度)は、検出器から与えられる応答信号によって決定されます。
それは比例します。つまり:

クロマトグラフィー定量の基礎:
定量分析の仕事は、混合サンプル中の何百もの成分を見つけることです
断片的なコンテンツ。クロマトグラフィーによる定量化は、以下に基づいて行われました: 操作条件が一貫している場合、
測定された成分の質量(または濃度)は、検出器から与えられる応答信号によって決定されます。
それは比例します。つまり:
1. ピーク面積の測定方法
ピーク面積はクロマトグラムによって提供される基本的な定量データであり、ピーク面積の測定精度は定量結果に直接影響します。異なるピーク形状のクロマトグラフィー ピークには異なる測定方法が使用されました。
定量分析で冬の正確な値を見つけるのは困難です。
一方で、絶対注入量を正確に測定することは困難であるため、他方では、
ピーク面積はクロマトグラフィー条件に依存するため、値を測定するときはクロマトグラフィー ストリップを維持する必要があります。
同じことを行うことは不可能でも便利でもありません。そしてたとえ正しく理解できたとしても
統一された基準がなく、直接適用できないため、正確な値も同様です。
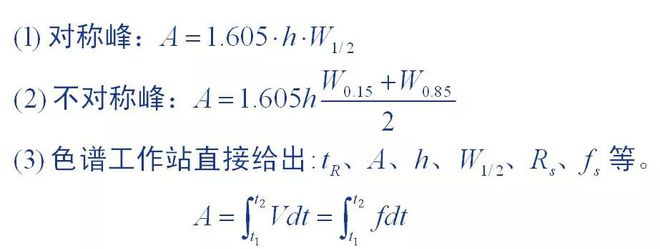
2.定量的補正係数
定量的補正係数の定義:検出器に入る成分の量(m)
クロマトグラフィーのピーク面積 (A) またはピーク高さ () の比は比例定数 (,
比例定数は、コンポーネントの絶対補正係数と呼ばれます。
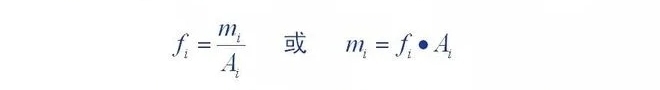
定量分析で冬の正確な値を見つけるのは困難です。
一方で、絶対注入量を正確に測定することは困難であるため、他方では、
ピーク面積はクロマトグラフィー条件に依存するため、値を測定するときはクロマトグラフィー ストリップを維持する必要があります。
同じことを行うことは不可能でも便利でもありません。そしてたとえ正しく理解できたとしても
統一された基準がなく、直接適用できないため、正確な値も同様です。
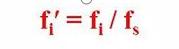
つまり、コンポーネントの相対補正係数はコンポーネントと基準物質です。
絶対補正係数の比率。
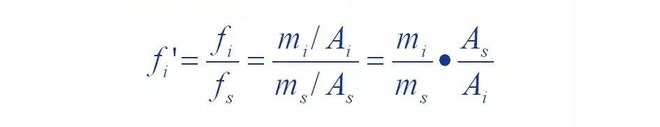
相対補正係数は、標準に対するコンポーネントの品質であることがわかります。
物質 s が等しい場合、標準物質のピーク面積は成分のピーク面積となります。
複数。ある成分の質量が m、ピーク面積が A の場合、f'A の数は次のようになります。
値は、質量を持つ標準物質のピーク面積に等しくなります。言い換えると、
相対補正係数により、各成分のピーク面積を分離できます。
標準物質のピーク面積とその質量に換算したときの比
規格が統一されています。これは、各コンポーネントの割合を計算するための正規化された方法です。
量の基本。
相対補正係数の取得方法: 相対補正係数の値を比較するだけです。
測定は標準と検出器のタイプに関係しますが、操作ストリップにも関係します
それは問題ではありません。したがって、値は文献の参考文献から取得できます。テキストの場合
オファリングに希望の価値が見つからない場合は、自分で決定することもできます。決定方法
方法:測定物質を一定量、選択した標準物質10個→一定濃度にする
2 つの成分のクロマトグラフィーのピーク面積 A および As を測定しました。
それが公式です。
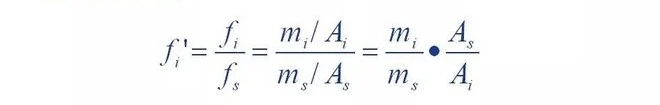
3. 定量的な計算方法
(1) 面積正規化方法
すべてのピークのない画分の含有量の合計を 100% として計算し、定量化しました。
この方法は正規化と呼ばれます。その計算式は次のとおりです。
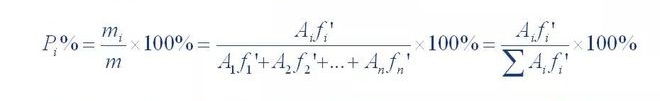
ここで、P,% は試験された成分の含有率です。A1、A2...A n は成分 1 です。1~n のピーク面積。f'1、f'2...f'n はコンポーネント 1 ~ n の相対補正係数です。
(2) 外部標準法
サンプル中の試験対象成分の応答信号と、対照としての純粋な試験対象成分とを定量的に比較する方法。
(3) 内部標準法
いわゆる内部標準法とは、被検物質の標準液や試料溶液に内部標準として純物質を一定量添加し、分析・定量する方法です。
(3)標準添加方法
標準添加法は内部添加法とも呼ばれ、(△C)を一定量添加する方法です。
被験物質の基準を被験サンプル溶液に添加し、試験をアッセイに追加した
物質を添加した後の試料溶液のピークが元の試料溶液のピークよりも高かった
面積の増分(△A)を用いて試料溶液中の物質の濃度を計算
コンテンツ (Cx)
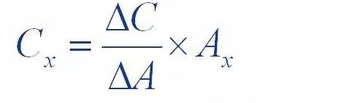
ここで、Ax は元のサンプル内の測定対象物質のピーク面積です。
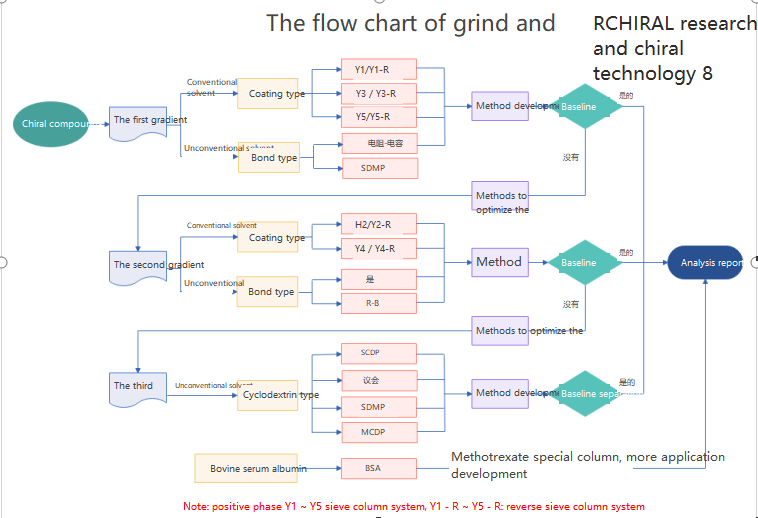
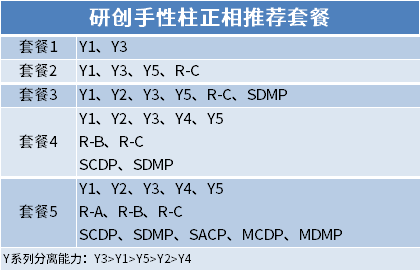

投稿日時: 2023 年 3 月 27 日